
- Fortran95
- C++
- MPI
- CUDA
Key words
- electronic transport
- electron-phonon interaction
- Anderson localization
- non-equilibrium Green's function
- density functional theory
Introduction
The Inelastic DISORDER code was developed for computing, from first principles methods, the electronic transport properties of 1-dimensional devices with a large number of randomly distributed defects considering the inelastic electron-phonon scattering. It combines electronic structure calculations via density functional theory (DFT) with the non-equilibrium Green’s function formalism (NEGF) for the transport.
The electron-phonon interaction is considered to be weak and localized in the defects. Under these circumstances, the current and power expressions can be expanded to second order in the electron-phonon couplings to obtain the so-called Lowest Order Expansion [1, 2]. Approximating the contact broadening and the non-interacting retarded Green’s functions as energy independent matrices (Wide-Band Limit), the current and power expressions take a simplified form where the integration over energy are calculated analytically.
I-DISORDER uses the Hamiltonian and overlap matrices from DFT calculations performed by the SIESTA code [3, 4], the leads calculations from SMEAGOL code [5] and the electron-phonon coupling from POSITIVE VIBRATIONS code [7]. For computing the non-equilibrium Green’s function I-DISORDER uses a recursive method [8, 9].
Compilation
The compilation of the program is done using a Makefile which, in turn, uses a arch.make file provided by the user, containing the required information for compiling the code.
Several examples of arch.make files for different machine architectures are provided at Obj/archmake folder.
Basically, in the arch.make file the user should set the C and Fortran compilers and its options, and should provide the path for BLAS [10] and LAPACK [11] libraries.
For parallel computing with Message Passing Interface MPI [12], the user should choose a MPI Fortran compiler and provide the path for MPI libraries.
If one wants to use CUDA-enable graphics processing units (GPU) for a GPU-accelerated computing, one should also provide the path for MAGMA [11], CUDA [14] and CUBLAS [15] libraries.
The code compilation takes place at Obj directory, which will hold the resulting object files, modules files and libraries.
In order to prepare the building of the code, one should go to Obj directory and run a script as follows:
sh ../Src/obj_setup.sh
Once the set-up is done, to proceed with the compilation just type:
make
This will create an executable file, as defined at arch.make file with the EXEC variable.
The System Set-Up
A typical disordered system simulated by I-DISORDER is presented in Figure 1. It comprises two semi-infinite leads (left and right) and an scattering region with randomly distributed defects.
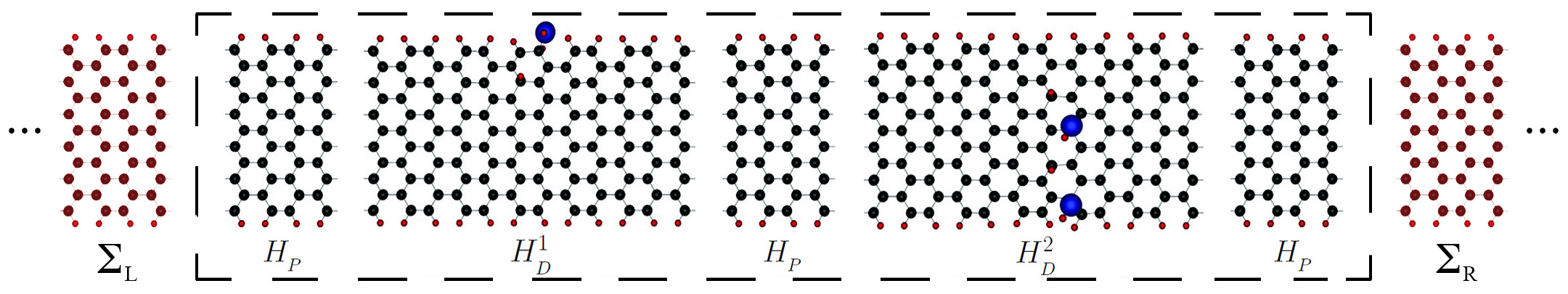
Figure 1: Representation of a typical disordered system using a graphene nanoribbon as an example. One identifies a scattering region with three pristine segments (HP) and two with defects (H1D and H2D). The left and right leads, made by semi-infinite pristine nanoribbons, are coupled to the first and last segments, respectively.
Before running I-DISORDER there are two preliminary steps: 1. the electronic structure calculating of each type of block that composes the system; 2. the vibrational calculation of each block with electron-phonon interaction.
The first step is carried out with SMEAGOL code with a lead-type computation (refer to SMEAGOL User’s Guide to see how to perform this calculation [6]). For each type of block, an electronic structure calculation is performed with the segment periodically repeated in the transport direction. The resulting Hamiltonian and overlap matrices from each unit cell are stored, as well as their respective couplings.
One must take a special care when building the blocks containing defects. They must have at least one pristine segment at each end to ensure that the coupling between two blocks is always equal to the coupling between pristine blocks.
The second step is to compute the vibrational modes and frequencies and the electron-phonon coupling matrices of each block with inelastic scattering. This is carried out with POSITIVE VIBRATIONS code (refer to POSITIVE VIBRATIONS User’s Guide to see how to perform this calculation [7]).
Input Files
From a lead-type computation with SMEAGOL code, the following files are generated: BlockLabel.HSL, bulklft.DAT and bulkrgt.DAT.
For the blocks that will constitute the leads one should copy all these files to the directory where the calculation with I-DISORDER will be performed and duplicate the file bulklft.DAT as LeadLabel.DAT.
For the other blocks one should copy the file BlockLabel.HSL and copy the file bulklft.DAT as BlockLabel.DAT.
From the vibrational calculation with POSITIVE VIBRATIONS code, one should copy only the file BlockLabel.Meph, which contains all calculated frequencies and electron-phonon coupling matrices.
Input Flags
The following input flags must be supplied to the input file (say input.in) in order to perform the transport calculation.
If any of these values is not supplied, the default value will be assumed.
SystemLabel (String): A single word (maximum of 60 characters without blanks) containing a nickname of the system, used to name output files.
Default value:
i-disorder
NumberUnits (Integer): Total number of blocks in the scattering region (in the example of Figure 1 the
NumberUnitsis 5).Default value:
1
NumberUnitTypes (Integer): Number of different types of blocks in the scattering region (in Figure 1 the
NumberUnitTypesis 3, since there are two different types of defects and the pristine block).Default value:
2
NumberDefects (Integer): Number of defects in the entire system (in Figure 1 the
NumberDefectsis 2). Note that this number should be <=NumberUnits.Default value:
1
UnitsFiles (data block): Block containing information about all different types of blocks. It contains
NumberUnitTypes+ 2 rows and 3 columns. The last 2 rows correspond to the left and right leads (in this order) and the antepenultimate (third last) correspond to the pristine block. The firstNumberUnitTypesrows correspond to the different types of blocks in the scattering region. The first column consists of real numbers which indicates the probability that the system contains each kind of block (the last three, from the leads and pristine block, are ignored). Note that the sum of the elements of the first column must be equal to 1. The second column contains an integer0or1indicating if there will be (1) or not (0) electron-phonon interaction in the block. The last column are strings the with block labels, which are used to find the filesBlockLabel.HSLandBlockLabel.DAT.Example:
%block UnitsFiles 0.5 1 Defect1 0.3 0 Defect2 0.2 1 Defect3 0.0 0 pristine 0.0 0 leftLead 0.0 0 rightLead %endblock UnitsFilesIn this example, the
NumberUnitTypesis 4 and there are 3 types of defects, with the first one with a higher probability of being present in the system. The electron-phonon interaction will be considered only on first and third defects blocks and the program will search for the filesDefect1.MephandDefect3.Meph.Default value: No Default
SpinPolarized (Boolean): If
SpinPolarizedis set toTit will be considered 2 spin components when reading the input files.Default value:
F
ElectronicTemperature (Physical): Temperature for Fermi-Dirac distribution.
Default value:
300.0 K
VInitial (Physical): Value of the initial bias.
Default value:
0.0 eV
VFinal (Physical): Value of the final bias.
Default value:
0.1 eV
NIVPoints (Integer): Number of bias steps considered between the two limits
VInitialandVFinal. The current and the dissipated power will be calculated only for these biases.Default value: 10
TransmInitial (Physical): Value of the initial energy.
Default value: -1.0 eV
TransmFinal (Physical): Value of the final energy.
Default value: 1.0 eV
NumberTransmPoints (Integer): Number of energy points uniformly distributed between
TransmInitialandTransmFinalfor which the current and the dissipated power are calculated.Default value: 100
AsymmGridPts (Integer): Number of energy grid points for computing the integral from asymmetric term of the inelastic current in LOE approximation.
Default value: 1000
PhononEquilibrium (Boolean): If
PhononEquilibriumis set toTthe phonons will be considered in thermodynamic equilibrium and will be described by the Bose-Einstein distribution. Otherwise an non-equilibrium heating of the phonon system will be considered.Default value:
T
PhononDamping (Physical): External damping rate. It is a phenomenological parameter related to the inverse of phonon’s lifetime and is used when considering non-equilibrium heating of the phonon system. If
PhononEquilibriumis set toT, this flag is ignored.Default value: 0.05 eV
GPU.ProcsPerGPU (Integer): Number of processes running in each GPU (in case I-DISORDER has been compiled with CUDA-enable graphics processing units for GPU-accelerated computing).
Default value: 1
Output Files
SystemLabel_ExVxI.CUR: File with six columns containing the computed currents. The first column corresponds to the energy in eV (subtracted by the Fermi energy) and the second column corresponds to the bias in V. The next columns are the computed current in ampere A with the following order: elastic current, symmetric term of inelastic current, asymmetric term of inelastic current and the total current.
SystemLabel_ExVxdI.dIdV: File with six columns containing the computed differential conductances dI/dV. The first column corresponds to the energy in eV (subtracted by the Fermi energy) and the second column corresponds to the bias in V. The next columns are the computed dI/dV in quantum conductance units (G0) with the following order: elastic dI/dV, symmetric term of inelastic dI/dV, asymmetric term of inelastic dI/dV and the total dI/dV.
SystemLabel_ExVxd2I.d2IdV2: File with six columns containing the computed derivatives d2I/dV2. The first column corresponds to the energy in eV (subtracted by the Fermi energy) and the second column corresponds to the bias in V. The next columns are the computed d2I/dV2 in quantum conductance per volt (G0V-1) with the following order: elastic d2I/dV2, symmetric term of inelastic d2I/dV2, asymmetric term of inelastic d2I/dV2 and the total d2I/dV2.
SystemLabel_ExVxP_nU.PWR: File containing the computed dissipated power by the block
nUwith electron-phonon interaction. The first column corresponds to the energy in eV (subtracted by the Fermi energy) and the second column corresponds to the bias in V. The following columns corresponds to the dissipated power in eV by each vibrational mode. The last column corresponds to the total dissipated power in eV (all modes).
SystemLabel_nU.SPCTR: File containing the computed spectral function of the block
nUwith electron-phonon interaction. The file indexed withnU+1(the number of the blocks with electron-phonon interaction plus 1) corresponds to the spectral function of all blocks with electron-phonon interaction. The first column corresponds to the energy in eV (subtracted by the Fermi energy) and the second column corresponds to the spectral function in (eV)-1.
SystemLabel_nU.DOS: File containing the computed local density of states (LDOS) of the block
nUwith electron-phonon interaction. The file indexed withnU+1(the number of the blocks with electron-phonon interaction plus 1) corresponds to the sum of the LDOS all blocks with electron-phonon interaction. The first column corresponds to the energy in eV (subtracted by the Fermi energy) and the second column corresponds to the LDOS in (eV)-1.
Running I-DISORDER
Assuming that the user has already compiled I-DISORDER successfully and saved it as an executable (say i-disorder) and assuming that the user has already performed the two preliminary steps described in The System Set-Up above, then we are ready to start a calculation.
The input files must be copied to the directory where the calculation will be performed, as described in Input Files above, and an file (say input.in) containing the input flags described in Input Flags above must also be present.
Then the user can run I-DISORDER by typing:
i-disorder < input.in > output.out
In case of parallel computing with $PROCS processes, the user can type:
mpirun -np $PROCS i-disorder < input.in > output.out
The I-DISORDER code will generate a system with a randomly distributed defects accordingly to the user’s options in input.in.
The resulting output files can be processed using a variety of plotting programs such as xmgrace for 2D-plots and OpenDX for 3D-plots and isosurfaces or gnuplot for either cases.
Some scripts for plotting the results with gnuplot can be find at Scripts/plot directory.
Usually, one is interested in running I-DISORDER code a large number of times and then compute the average values.
For that, the user will find some useful scripts at Scripts/analysis and Scripts/submit directories.
Problem Handling
The users are encouraged to report problems and bugs to the I-DISORDER’s developers at github. Patches and fixes will be uploaded to the web-site https://github.com/brandimarte/idisorder.
Acknowledgements
PBM thanks CNPq (National Council of Technological and Scientific Development) for financial support (grant 142792/2010-1).
References
[1] J. K. Viljas, J. C. Cuevas, F. Pauly and M. Häfner, Phys. Rev. B 72, 245415 (2005).
[2] M. Paulsson, T. Frederiksen and M. Brandbyge, Phys. Rev. B 72, 201101(R) (2005).
[4] http://departments.icmab.es/leem/siesta/
[6] SMEAGOL User’s Guide comes along with the code at: http://www.smeagol.tcd.ie
[7] PhOnonS ITeratIVE VIBRATIONS. https://github.com/brandimarte/vibrations
[8] C. H. Lewenkopf and E. R. Mucciolo, J. Comput. Electron. 12, 203-231 (2013).
[9] A. R. Rocha, M. Rossi, A. J. R. da Silva and A. Fazzio, J. Phys. D: Appl. Phys. 43, 374002 (2010).
[10] BLAS - Basic Linear Algebra Subprograms. http://www.netlib.org/blas/
[11] LAPACK - Linear Algebra PACKage. http://www.netlib.org/lapack/
[12] M. Snir, S. Otto, S. Huss-Lederman, D. Walker and J. Dongarra, MPI: The Complete Reference, The MIT Press (1996).
[13] MAGMA - Matrix Algebra on GPU and Multicore Architectures. http://icl.cs.utk.edu/magma/
[14] NVIDIA CUDA Parallel Programming and Computing Platform. http://www.nvidia.com/object/cuda_home_new.html
[15] CUBLAS - NVIDIA CUDA Basic Linear Algebra Subroutines. https://developer.nvidia.com/cublas